
La sphérocytose héréditaire est une maladie génétique du sang qui fragilise les globules rouges. Elle reste la cause la plus fréquente d’anémie hémolytique héréditaire dans les populations d’origine nord-européenne, avec une fréquence estimée entre 1 personne sur 2 000 et 1 sur 5 000. Une étude menée en Europe centrale a permis d’identifier un nombre inhabituellement élevé de nouveaux variants génétiques responsables de la maladie, avec des conséquences concrètes pour le diagnostic et le suivi des familles concernées. Cet article fait le point sur les causes, les symptômes, le diagnostic et les traitements de la sphérocytose héréditaire, à la lumière de ces avancées récentes.
Qu’est-ce que la sphérocytose héréditaire ?
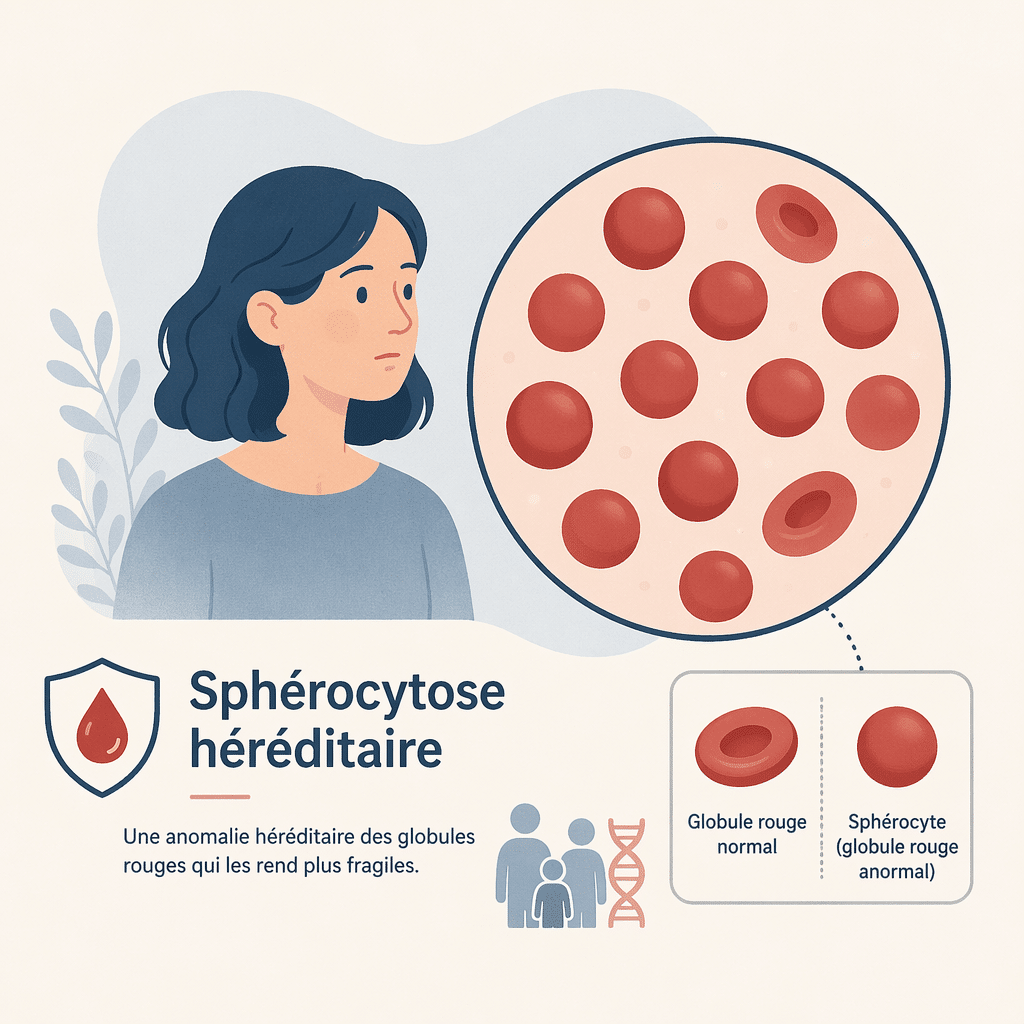
La sphérocytose héréditaire, autrefois appelée maladie de Minkowski-Chauffard, est une maladie génétique qui touche la membrane des globules rouges. Normalement, un globule rouge a la forme d’un disque légèrement creux sur chaque face, ce qui lui permet de se déformer facilement pour circuler dans les plus petits vaisseaux sanguins. Dans la sphérocytose, un défaut de la membrane rend les globules rouges plus rigides et plus petits : on parle de « sphérocytes », d’où le nom de la maladie.
Ces sphérocytes, plus fragiles, sont repérés et détruits prématurément par la rate. Cette destruction accélérée, appelée hémolyse, entraîne une anémie (baisse du nombre de globules rouges), une jaunisse et, chez la plupart des patients, une augmentation du volume de la rate (splénomégalie). La sphérocytose héréditaire touche aussi bien les hommes que les femmes et peut se manifester à tout âge, même si elle est souvent découverte dès la période néonatale ou dans l’enfance.
Les causes génétiques de la maladie
La sphérocytose héréditaire résulte de mutations — des changements dans un gène qui modifient la protéine qu’il code — touchant l’un des cinq gènes responsables de la fabrication des protéines de la membrane du globule rouge : ANK1 (ankyrine-1), SPTB (bêta-spectrine), SPTA1 (alpha-spectrine), SLC4A1 (protéine de la bande 3, qui transporte certains ions) et EPB42 (protéine 4.2). Ces protéines forment un échafaudage sous la membrane du globule rouge ; lorsque l’une d’elles est déficiente, cet échafaudage perd de sa cohésion et le globule rouge devient sphérique et fragile.
La transmission se fait le plus souvent sur un mode autosomique dominant : environ 75 % des personnes atteintes ont hérité de la maladie d’un seul de leurs deux parents, qui a lui-même une chance sur deux de transmettre le gène muté à chacun de ses enfants. Dans une minorité de cas, la transmission est récessive (les deux parents transmettent chacun une copie du gène altéré) ou résulte d’une mutation apparue spontanément chez l’enfant, sans antécédent familial (mutation de novo).
Symptômes et manifestations cliniques
La sévérité de la sphérocytose héréditaire varie énormément d’une personne à l’autre, y compris au sein d’une même famille. Certains patients restent asymptomatiques pendant des années, tandis que d’autres présentent des symptômes marqués dès la naissance.
La triade classique
Trois manifestations reviennent fréquemment :
La jaunisse (ictère) : chez le nouveau-né, elle est souvent le premier signe visible. Elle correspond à l’accumulation de bilirubine, un pigment jaune issu de la dégradation de l’hémoglobine libérée par les globules rouges détruits. Une jaunisse néonatale sévère peut nécessiter une photothérapie pour éviter tout risque cérébral.
L’anémie : elle se traduit par une fatigue, un essoufflement à l’effort, une pâleur ou des vertiges. Sa sévérité dépend du degré d’hémolyse et de la capacité de la moelle osseuse à compenser la destruction des globules rouges.
La splénomégalie : l’augmentation du volume de la rate, qui filtre et élimine les sphérocytes les plus fragiles, peut provoquer une sensation de pesanteur abdominale.
Complications possibles
Certaines complications peuvent survenir au cours de l’évolution de la maladie, notamment des calculs biliaires (lithiase vésiculaire) liés à l’excès de bilirubine, ou des crises aplasiques : des épisodes d’arrêt temporaire de la production de globules rouges, souvent déclenchés par une infection au parvovirus B19, qui peuvent provoquer une anémie brutale nécessitant parfois une transfusion.
Diagnostic de la sphérocytose héréditaire
Le diagnostic repose sur un faisceau d’éléments : les antécédents familiaux, l’examen clinique et plusieurs analyses de sang. La numération formule sanguine (NFS) met en évidence l’anémie et permet d’évaluer les réticulocytes (jeunes globules rouges), souvent élevés en réponse à l’hémolyse. L’examen du frottis sanguin au microscope révèle la présence de sphérocytes, un indice important mais non suffisant à lui seul pour confirmer le diagnostic.
Des tests plus spécifiques permettent de confirmer la maladie et d’éliminer d’autres causes d’anémie hémolytique, comme la cytométrie en flux après marquage à l’éosine-5-maléimide (test EMA), aujourd’hui recommandée en première intention car simple à réaliser et disponible dans de nombreux laboratoires. L’ektacytométrie en gradient osmolaire, plus rarement disponible en France, reste l’examen de référence pour les cas complexes.
Une analyse génétique, recherchant une mutation dans l’un des cinq gènes connus, peut être proposée en complément, en particulier lorsque le tableau clinique est atypique, en l’absence d’antécédents familiaux, chez le nouveau-né, ou avant d’envisager une splénectomie afin d’écarter formellement une stomatocytose héréditaire (une autre maladie de la membrane du globule rouge, qui contre-indique cette intervention).
Sévérité selon le type génétique : ce que l’on sait aujourd’hui
Un des apports importants des travaux récents est la mise en évidence de liens entre le gène affecté, le type de mutation et la sévérité clinique observée chez les patients. Ce tableau résume les grandes tendances rapportées dans plusieurs cohortes, à titre indicatif : la sévérité réelle dépend toujours du profil génétique complet de chaque patient et doit être évaluée individuellement par un médecin.
| Gène concerné | Fréquence observée | Tendance de sévérité rapportée |
|---|---|---|
| ANK1 | Gène le plus souvent en cause dans plusieurs cohortes européennes et nord-américaines | Variable, dépend du type de mutation ; nombreuses mutations à fort impact (décalage du cadre de lecture, arrêt prématuré) |
| SPTB | Deuxième gène le plus fréquemment touché | Variable, souvent des formes modérées |
| SLC4A1 | Présent chez une minorité de patients selon les cohortes | Formes hétérozygotes souvent plus douces ; formes homozygotes (les deux copies du gène touchées) associées à une sévérité accrue |
| SPTA1 | Gène le moins fréquemment en cause | Formes le plus souvent modérées, transmission parfois récessive |
Dernières avancées scientifiques
Une équipe de recherche autrichienne a mené la première étude portant spécifiquement sur le profil génétique de la sphérocytose héréditaire dans une population d’Europe centrale. Selon cette étude, publiée dans la revue HemaSphere, les chercheurs ont analysé 113 personnes issues de 35 familles, dont 69 patients atteints et 44 proches non affectés, à l’aide du séquençage de nouvelle génération — une technique qui permet de lire en détail l’ADN à la recherche de mutations.
Une découverte marquante : sur les 34 variants génétiques différents identifiés chez les patients, plus de la moitié (19 variants) n’avaient jamais été décrits auparavant dans la littérature scientifique. Ce que cela change pour les patients et les familles : chaque nouvelle mutation répertoriée enrichit les bases de données génétiques utilisées par les laboratoires pour interpréter les résultats des tests. Concrètement, un patient dont le variant est déjà connu et documenté a plus de chances d’obtenir une réponse claire et rapide lors d’une analyse génétique, ce qui facilite le dépistage chez les autres membres de la famille et la planification du suivi médical.
L’étude a également confirmé qu’ANK1 restait le gène le plus souvent impliqué (chez un peu moins de la moitié des patients de la cohorte), suivi de SPTB, SLC4A1 puis SPTA1 — un classement globalement cohérent avec ce qui est observé dans d’autres régions du monde, avec toutefois des nuances régionales. Les chercheurs ont aussi mis en évidence un lien entre certains profils génétiques et la sévérité clinique : les patients porteurs de deux copies altérées du gène SLC4A1 (forme homozygote) présentaient des formes plus marquées de la maladie, tandis qu’une seule copie altérée (forme hétérozygote) s’accompagnait le plus souvent d’un tableau plus modéré.
Nuance de fiabilité : il s’agit d’une étude portant sur un nombre de patients relativement restreint, menée dans un centre unique. Les auteurs soulignent eux-mêmes que des cohortes plus larges et des études menées dans plusieurs centres seront nécessaires pour confirmer ces liens entre génotype et sévérité de façon plus robuste. Ces résultats représentent donc une avancée importante pour la compréhension de la maladie, mais ne bouleversent pas la prise en charge actuelle : ils viennent surtout affiner la précision du diagnostic génétique et l’anticipation de l’évolution clinique, sans remplacer le suivi clinique habituel.
Traitements et prise en charge
Il n’existe pas de traitement curatif de la sphérocytose héréditaire à proprement parler, mais plusieurs approches permettent de limiter l’hémolyse et ses conséquences.
Chez le nouveau-né, la jaunisse sévère est prise en charge par photothérapie. En cas d’anémie sévère et symptomatique, une transfusion de globules rouges peut être nécessaire, notamment lors des crises aplasiques. Une supplémentation en folates (vitamine B9) est généralement recommandée dans les formes modérées à sévères, car ces vitamines sont indispensables à la fabrication des nouveaux globules rouges.
La splénectomie (ablation de la rate) reste le seul geste qui réduit nettement l’hémolyse chronique, puisque la rate est le principal site de destruction des sphérocytes. Elle n’est cependant pas systématique : son indication dépend de la sévérité de l’anémie et de son retentissement clinique. Lorsqu’elle est envisagée, elle est généralement retardée jusqu’à l’âge de 5 à 6 ans en raison du rôle de la rate dans la défense contre certaines infections, en particulier chez le jeune enfant. Des vaccinations spécifiques (pneumocoque, méningocoque, Haemophilus influenzae B) et une antibioprophylaxie sont mises en place avant et après l’intervention. En cas de calculs biliaires symptomatiques, une ablation de la vésicule biliaire (cholécystectomie) peut être proposée, parfois associée à la splénectomie.
Quand consulter un médecin
Certains signes doivent inciter à consulter rapidement, en particulier chez un enfant déjà suivi pour une sphérocytose héréditaire ou chez un proche d’une personne atteinte :
- Une jaunisse qui apparaît ou s’aggrave, surtout chez le nouveau-né.
- Une fatigue inhabituelle, une pâleur marquée, un essoufflement ou des vertiges pouvant traduire une aggravation de l’anémie.
- Une fièvre chez une personne ayant subi une splénectomie, qui constitue une urgence en raison du risque infectieux accru après l’ablation de la rate.
- Des douleurs abdominales, en particulier en haut à droite du ventre, pouvant évoquer un calcul biliaire.
- Tout antécédent familial de sphérocytose héréditaire chez une personne présentant une anémie ou une jaunisse inexpliquée, qui justifie un bilan sanguin et un avis médical dédié.
Glossaire
- Mutation : changement dans la séquence d’un gène qui peut modifier ou empêcher la fabrication normale d’une protéine, ici une protéine de la membrane du globule rouge.
- Génotype : ensemble des caractéristiques génétiques d’une personne, ici le ou les variants responsables de la maladie.
- Phénotype : ensemble des manifestations cliniques observables chez un patient, qui peut varier même pour un même génotype.
- Hémolyse : destruction des globules rouges plus rapide que la normale.
- Sphérocyte : globule rouge devenu sphérique et rigide en raison d’un défaut de sa membrane, caractéristique de la sphérocytose héréditaire.
- Splénectomie : intervention chirurgicale consistant à retirer tout ou partie de la rate.
- Séquençage de nouvelle génération (NGS) : technique de laboratoire qui permet de lire rapidement de grandes portions d’ADN pour y rechercher des mutations.
- Transmission autosomique dominante : mode de transmission génétique où une seule copie altérée du gène, héritée d’un seul parent, suffit à provoquer la maladie.
- Réticulocytes : globules rouges jeunes et immatures, dont le nombre reflète l’activité de production de la moelle osseuse.
- Bilirubine : pigment jaune issu de la dégradation de l’hémoglobine, responsable de la jaunisse lorsqu’il s’accumule dans le sang.
Foire aux questions
La sphérocytose héréditaire se transmet-elle toujours des parents aux enfants ?
Pas toujours. Dans environ 75 % des cas, elle se transmet sur un mode autosomique dominant : un seul parent porteur du gène muté suffit à transmettre la maladie, avec un risque de transmission à chaque enfant d’environ une chance sur deux. Dans une minorité de cas, la transmission est récessive (les deux parents transmettent chacun une copie altérée) ou la mutation apparaît spontanément chez l’enfant, sans antécédent familial connu.
Quels sont les principaux symptômes de la sphérocytose héréditaire ?
Les trois signes les plus fréquents sont la jaunisse, l’anémie et l’augmentation du volume de la rate. Leur intensité varie beaucoup d’une personne à l’autre : certains patients n’ont presque aucun symptôme, tandis que d’autres présentent une anémie plus marquée nécessitant un suivi rapproché, voire des transfusions ponctuelles.
Comment se pose le diagnostic de sphérocytose héréditaire ?
Le diagnostic associe l’examen clinique, les antécédents familiaux et des analyses de sang, notamment la numération formule sanguine et l’examen du frottis sanguin. Un test de cytométrie en flux (test EMA) permet de confirmer le diagnostic dans la grande majorité des cas. Une analyse génétique peut compléter le bilan, en particulier en cas de doute diagnostique ou avant une splénectomie.
Quel est le traitement de la sphérocytose héréditaire ?
Il n’existe pas de traitement curatif de la maladie elle-même, mais la prise en charge repose sur la gestion des symptômes : supplémentation en folates, transfusions en cas d’anémie sévère, et surveillance régulière. La splénectomie, lorsqu’elle est indiquée, réduit nettement l’hémolyse chronique, mais son indication est évaluée au cas par cas par l’équipe médicale.
Les nouveaux variants génétiques découverts changent-ils le traitement ?
Pas directement. Ces découvertes affinent surtout la précision du diagnostic génétique et la compréhension du lien entre le gène affecté et la sévérité de la maladie. Elles n’entraînent pas de changement immédiat des traitements disponibles, mais elles peuvent, à terme, aider les médecins à mieux anticiper l’évolution clinique de chaque patient.
La sphérocytose héréditaire peut-elle passer inaperçue pendant longtemps ?
Oui. Certaines formes légères ou compensées ne provoquent aucun symptôme notable pendant des années, et la maladie est parfois découverte à l’occasion d’un bilan sanguin de routine, d’une grossesse, ou d’une crise aplasique déclenchée par une infection virale. C’est pourquoi un antécédent familial connu justifie souvent un dépistage, même en l’absence de symptômes.
Sources
- Haute Autorité de Santé (HAS) — Protocole national de diagnostic et de soins : Sphérocytose héréditaire et autres anémies hémolytiques par anomalie de la membrane érythrocytaire
- Orphanet — Sphérocytose héréditaire
- Filière de santé maladies rares MCGRE (CHU Bicêtre, AP-HP) — Fiche d’informations patient : la sphérocytose
- Kager L., Jimenez-Heredia R., Segarra-Roca A. et al. — « A single-center cohort study of patients with hereditary spherocytosis in Central Europe reveals a high frequency of novel disease-causing genotypes » — HemaSphere, 2024 — https://doi.org/10.1002/hem3.31
Autres articles pour aller plus loin
- Anémie : causes, symptômes, diagnostic et traitements
- Réticulocytes : le guide complet
- Haptoglobine : comprendre ce marqueur sanguin
- Bilirubine totale : explications claires
- Maladie des agglutinines froides : le traitement sutimlimab
Comprendre une numération formule sanguine, un taux de réticulocytes ou de bilirubine peut sembler complexe, en particulier lorsqu’une maladie génétique comme la sphérocytose héréditaire est suspectée ou déjà suivie dans la famille. Ces résultats prennent tout leur sens lorsqu’ils sont mis en perspective les uns par rapport aux autres, sans pour autant remplacer l’avis d’un professionnel de santé. AI DiagMe vous aide à décrypter vos analyses de laboratoire en langage clair, pour mieux préparer vos échanges avec votre médecin.
Interprétez vos analyses de laboratoire avec AI DiagMe
Obtenez une interprétation en quelques minutes


{kind=link}
{kind=link}
{kind=link}